ISOX-DUAL Derived PROTACs and their Collapse
A look at some recent research in the field of targeted protein degradation: a collaborative effort led by John Spencer at the University of Sussex and Hannah Maple at Tocris with crucial protein structure input by Dr Andreas Joerger (Frankfurt). Using x-ray structure guided design to modify the CBP/BRD4 inhibitor ISOX-DUAL led to a series of dual-targeting proteolysis targeting chimeras (PROTACs) with high affinities. In the process of studying these compounds interesting cases of ‘degrader collapse’ was observed, suggesting an underappreciated mechanism by which targeted protein degraders fail to produce meaningful activities.
The c-Myc oncoprotein is a transcription factor implicated in more than 70% of cancers and is considered a good therapeutic target for treatments. However this is hindered by a lack of binding site for potential drug molecules, so approaches often focus on upstream proteins such as BRD4 and IRF4. In the case of IRF4, its long half-life means that even when expression of IRF4 is reduced through inhibition of the CBP/EP300 complex, protein levels are not significantly reduced. With this in mind, the authors thought a degrader approach might be effective: particularly as PROTACs are known to reduce protein concentrations long after the degrader itself has become undetectable. The fact that BRD4 and CBP/EP300 both contain bromodomains, also means such a degrader might show synergistic effects. By using ISOX-DUAL, a known μM inhibitor of both proteins, as the basis of the targeting ligand it could result in more effective reduction of c-Myc levels.
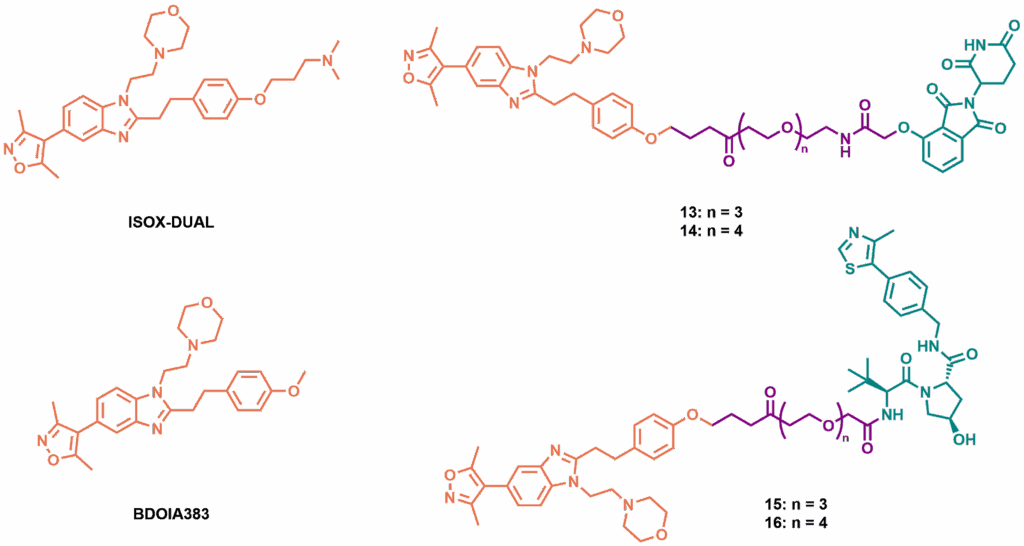
Structures of dual CBP/BRD4 targeting inhibitors ISOX-DUAL and BDOIA383 and range of phenyl ether derived PROTAC analogues, featuring CRBN-recruiting ligand (top) and VHL-recruiting ligand (bottom)
By examining published crystal structures of BDOIA383 (a methoxy analogue of ISOX-DUAL) with both BRD4 and CBP, two promising exit vectors for the attachment of E3 ligase ligands were identified: both the morpholine and phenyl ether remained solvent accessible in both crystal structures. Initial modifications to the ISOX-DUAL scaffold through the phenyl ether were tested for binding affinity by FRET assay, which revealed that longer linkages were the most promising, showing negligible loss of activity in biochemical assays. While these early analogues were used as initial materials to produce ISOX-DUAL phenolic ether degraders, an improved synthesis allowed the creation of a series of candidate PROTACs, with ligands to recruit both CRBN and VHL E3 ligases. Of these, FRET assays showed that most retained good affinity for BRD4, but all showed significant increases in their CBP IC50 values.
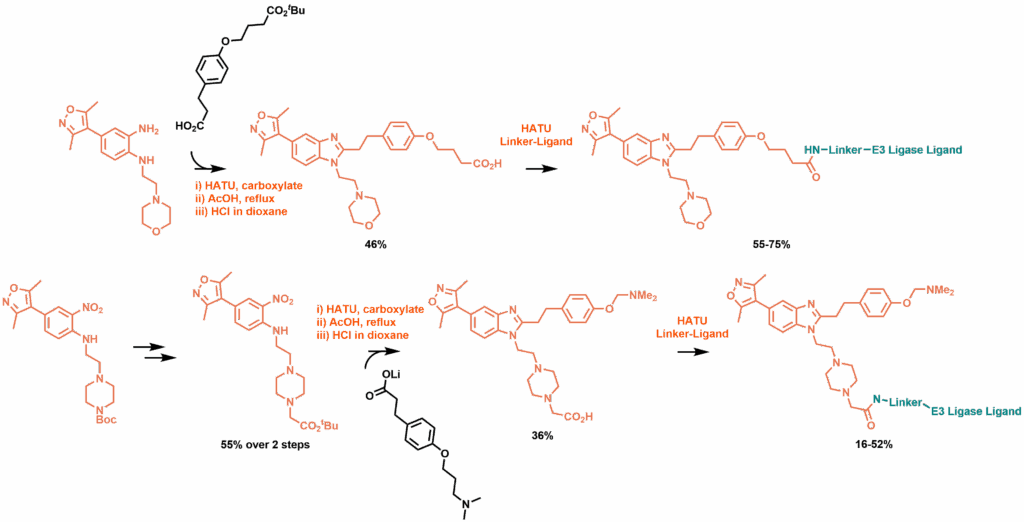
Synthetic routes to phenyl ether linked degraders (top) and piperazine linked degraders (bottom)
In exchanging the morpholine for a piperazine, the alternative viable attachment position could be pursued. An initial synthesis involving a problematic late-stage functionalisation of the piperazine was replaced by an improved method involving the early installation of an alpha tert-butyl ester. Again, FRET assays of early modifications suggested that longer linkages were required to avoid loss of affinity to both CBP and BRD4, and from here a large range of piperazine linked degraders were synthesised, again targeting both CRBN and VHL. Among these, all showed a drop in affinity to CBP and increased affinity for BRD4, despite the earlier results suggesting that the piperazine was a preferable route of attachment.
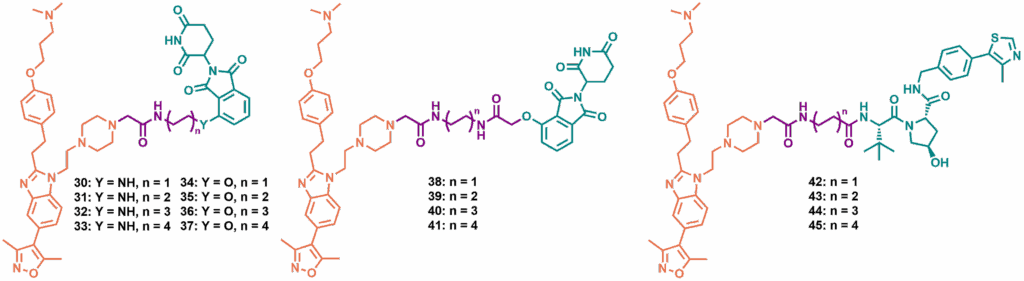
Range of piperazine-linked targeted protein degraders synthesised, including CRBN-recruiting 30-41 and VHL-recruiting 42-45
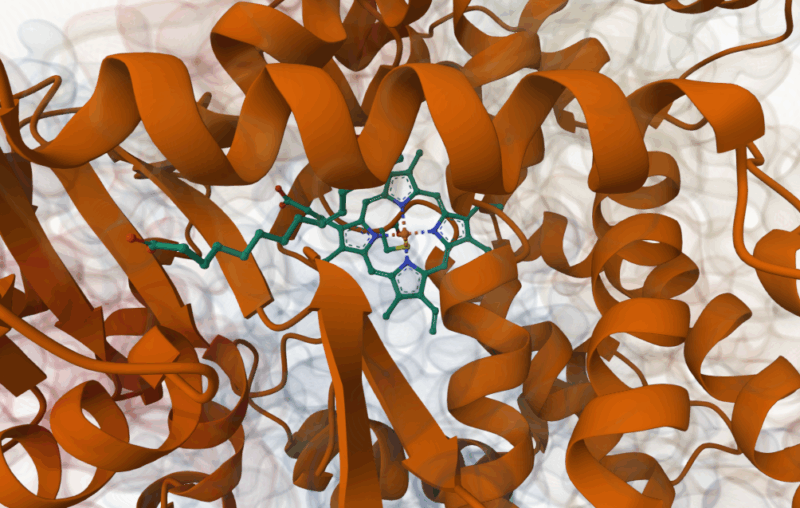
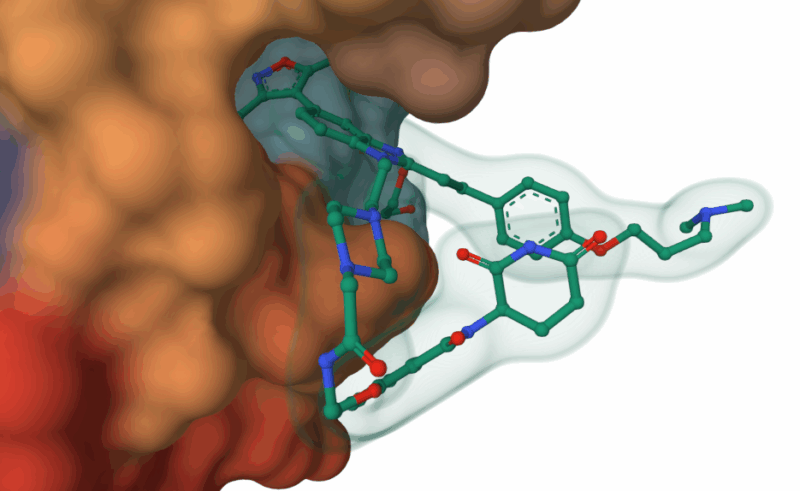
The VHL-targeting PROTACs 42-45 were tested in vitro, BRD4 was shown to be ubiquitinated, but no effect on CBP was observed. Similarly the CRBN-targeting molecules 34-36 were also tested in vitro: again, CBP was not ubiquitinated and only those compounds with the longest linkers showed activity against BRD4. Crystal structures of representatives of both the phenolic ether and piperazine degraders bound to BRD4 offered an insight into this variation in activity at odds with the apparent binding affinities. The phenyl-linked degrader 14 can be seen bound in a similar fashion to its parent compound BD0IA383, with the thalidomide CRBN ligand remaining solvent exposed. However, the piperazine linked analogue 34 is rotated 180 degrees within the pocket, and importantly, the protein itself is distorted with the thalidomide folded back along its surface. This ‘collapse’ of the degrader’s conformation, if maintained in solution, would explain the high affinity for BRD4, but low levels of ubiquitination. This mechanism could be an explanation for other reported ‘mismatches’ between PROTACs’ affinity and activity. The nanomolar activity of 34 (IC50 = 65 nM) is similar to potent BRD4 inhibitor (+)-JQ-1, and comparison of binding modes of the two compound showed some similarities, with both interacting with a flipped Trp81 residue. The co-crystal structures of VHL-based PROTACs 44 & 45 consistently showed a disordered and solvent exposed VHL ligand, explaining its ability to ubiquitinate BRD4. The linker itself was often adjacent to a hydrophobic surface of the protein, possibly explaining its increased affinity for BRD4 over the parent compound.
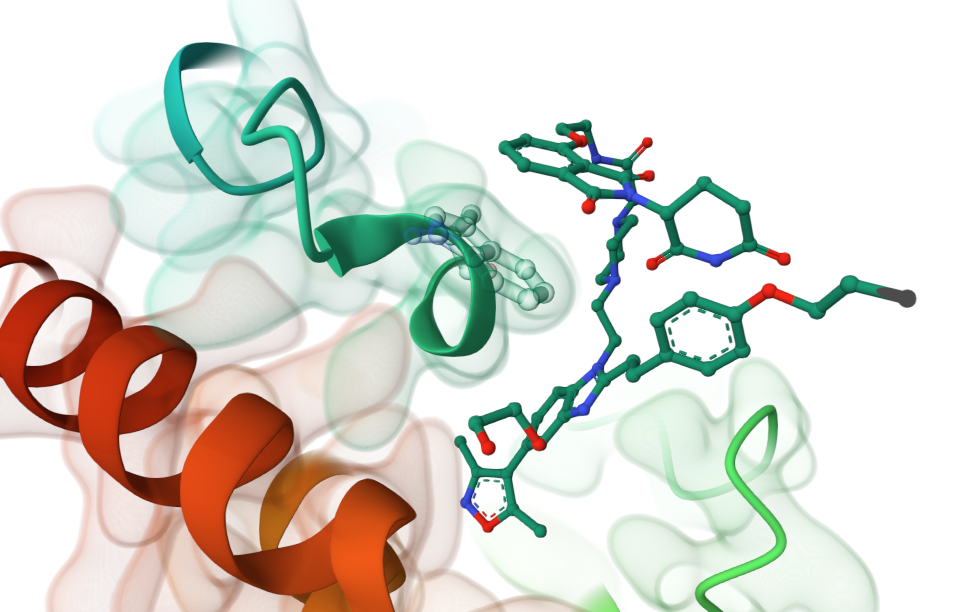
Piperazine-linked targeted protein degrader 34 co-crystallised with BRD4, showing collapse of structure and CRBN ligand lying across hydrophobic residue Trp81 (Balourdas, D.I., Edmonds, A.K., Marsh, G.P., Maple, H.J., Spencer, J., Knapp, S., Joerger, A.C.(2024) https://doi.org/10.2210/pdb9F1L/pdb)
These insights into the design of dual-targeting degraders highlight the importance of understanding the wider structural interactions of PROTAC binding, linker design, and the conformational effects that can lead to degrader collapse. While in this work, accurate degradation results were marred by the highly cytotoxic nature of the compounds, it opens the door to improved dual-degrader designs, with improved balance between protein targets through optimised design enhancing selectivity and activity.
Dr M. Tomsett – Technical Liason Officer
- Anthony K. Edmonds, Dimitrios-Ilias Balourdas, Graham P. Marsh, Robert Felix, Bradley Brasher, Jeff Cooper, Cari Graber-Feesl, Madhu Kollareddy, Karim Malik, Helen Stewart, Timothy J. T. Chevassut, Ella Lineham, Simon Morley, Oleg Fedorov, James Bennett, Mohan B. Rajasekaran, Samuel Ojeda, Drew A. Harrison, Christopher J. Ott, Andreas C. Joerger, Hannah J. Maple, and John Spencer, Journal of Medicinal Chemistry 202568 (9), 9638-9660
- Balourdas, D.I., Edmonds, A.K., Marsh, G.P., Maple, H.J., Spencer, J., Knapp, S., Joerger, A.C., Structural Genomics Consortium (SGC), First bromodomain of BRD4 in complex with ISOX-DUAL based degrader 35 (2024) https://doi.org/10.2210/pdb9F1L/pdb
- Eugene L. Piatnitski Chekler, Jessica A. Pellegrino, Thomas A. Lanz, R. Aldrin Denny, Andrew C. Flick, Jotham Coe, Jonathan Langille, Arindrajit Basak, Shenping Liu, Ingrid A. Stock, Parag Sahasrabudhe, Paul D. Bonin, Kevin Lee, Mathew T. Pletcher, Lyn H. Jones, Transcriptional Profiling of a Selective CREB Binding Protein Bromodomain Inhibitor Highlights Therapeutic Opportunities, Chemistry & Biology, Volume 22, Issue 12, 2015, 1588-1596